- Introduction
- Workflow
- DNA-derived Data Extension Terms for Metabarcoding Data
- DNA-derived Data Extension Terms for qPCR Data
- References
- Example Datasets
- Relevant R/Python packages
Introduction
DNA-derived biological occurrence data allow us to observe inconspicuous and cryptic taxa. DNA-derived data may come from sampling the environment (e.g. water, sediment), bulk organisms, or individual organisms. The derived data may be backed by preserved physical material or not, and may result from genetic sequencing or other DNA detection methods, such as qPCR.
Like all biological observation data, DNA-derived data should be as standardized and reproducible as possible, regardless of whether or not the detected species have formal scientific names. It is recommended practice that processed data should be provided as an extension to the occurrence core/extension in the DwC-A schema. The number of DNA-derived datasets in OBIS is limited (currently only 26) and our goal is to mobilize more datasets through this workshop. In this workshop we will focus on the two analytical methods that are typically used in studying environmental DNA (eDNA):
- Those which aim to detect a specific organism (qPCR/ddPCR)
- Those which describe an assemblage or community of organisms (metabarcoding).
qPCR/ddPCR
For the detection of specific species in eDNA-samples, most analyses include species-specific primers and qPCR (Quantitative Polymerase Chain Reaction) or ddPCR (Droplet-Digital Polymerase Chain Reaction). These methods do not generate DNA-sequences, and the occurrence data are completely dependent on the specificity of primers or assays used, so it is highly recommended that this information be preserved and shared with the data.
Metabarcoding
Metabarcoding utilizes general primers to recover hundreds to millions of DNA-sequences from a DNA extract. Sequences are typically identified and grouped, either by clustering sequences into operational taxonomic units (OTUs) or by denoising the data, which produces amplicon sequence variants (ASVs). The sequences that have been identified can be assigned a taxon by comparing them against reference databases, sometimes called reference libraries. To avoid confusion with the “libraries” that are the results of the sequencing process, we will try to refer to them as reference databases.
Archiving Raw Sequences
The raw, pre-processed sequences should be preserved in external archives or repositories (e.g. GenBank, ENA).
Taxonomic Assignment
A representative sequence for each OTU/ASV can be assigned a taxon by comparing them against reference databases. The efficacy of assignment depends on the completeness (coverage) ande reliability of reference libraries, as well as the tools used to carry out the classification. For example, many assignment algorithms are based on some kind of minimum confidence threshold and may result in OTUs/ASVs that are unassigned or badly assigned. These are all moving targets, making it essential to apply taxonomic expertise and caution in the interpretation of results.
It may be that you are only able to confidently assign your sequences a high-level classification, like Kingdom. However, it is still worth sharing standardized data and metadata so they can be re-analyzed and built upon as methods and reference databases improve. For genetic sequencing data to be reusable and reproducible, it is important to provide data users with information about the provanance and processing of the DNA. For example, habitat where the sample was collected from, DNA sequence, primers used, targeted gene, and referencing the sampling protocol . When applicable, providing the verified amplicon sequence variants (Сallahan et al. 2017) allow for precise reinterpretation of data, intra-specific population genetic analyses (Sigsgaard et al. 2019) and is likely to increase identification accuracy.
Workflow
The following workflow diagram is taken from the GBIF DNA-derived data manuscript. We are mainly focusing on the lower half of this diagram, particularly in standardizing genomic data to DwC and providing proper referencing to protocols and other metadata required for understanding the context in which the data was collected.

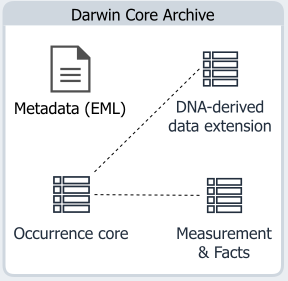
DNA-derived extension
The DNA-derived data extension is an extension in the DwC-A schema, and has to be used as an extension to Occurrence Core. It can be used in combination with other extensions, such as the Measurement & Facts extension. Information included in the DNA-derived extension should make it easier for data users to understand, analyze or reuse your genomic occurrence data. The value in adding your DNA-sequence or genomic data to GBIF comes from associating spatio-temporal occurrence data and DNA-based names from the data. It helps provide a mechanism for storing occurrence records of undescribed species. When these species become linked to a Linnean name, all these linked occurrence records will be immediately available. Including this data in GBIF increases citability, can hasten its discovery and integration in biological conservation and policy-making. Specific granularity is required for accurate reproducibility, especially in a field where protocols used can vastly impact the taxa observed.
5 Categories of DNA-derived data
Typically, molecular approaches to biodiversity characterization through qPCR and metabarcoding can be separated into 5 categories. Information collected in each of these categories can be included in the DNA-derived extension.
- Category 1 (metabarcoding): DNA-derived occurrences = DNA sequence or detection through PCR is the only evidence for presence of a given organism or community
- Category 2 (metabarcoding): Enriched occurrences = if some genetic materials are, or can be, associated with an observation or a specimen (not the only evidence of occurrences).
- Category 3 (qPCR): Targeted species detection (qPCR/ddPCR) = data where a specific (qPCR/ddPCR) assay is used to detect presence/absence of DNA sequence specific to target organism.
- Category 4 (metabarcoding): Name references (e.g. iBOL) = DNA-derived names, derived from clustering (OTU) or denoising (ASV)
- Category 5 (metabarcoding): Metadata-only datasets
See for a decision tree Table 1. For examples of datasets in GBIF for each of these categories see sections 2.1.1. - 2.1.5.
Tables
The DNA-derived data extension will have different requirements based on the analytical method applied in your project (i.e. metabarcoding vs. qPCR). Although few fields of the DNA-derived extension are “required” for metabarcoding and qPCR data, the decision to exclude “highly recommended” fields could severely impact how reusable your data will be to others. Therefore, when dealing with metabarcoding or qPCR data, it is (highly) recommended that aside from the general required fields in DwC the following (free-text) fields are also included:
Table 1. Recommended fields for Occurrence core or extension. An extended version of this table is found in GBIF manual.
| Metabarcoding | qPCR | ||
|---|---|---|---|
| Darwin Core Term | Description | Description | Required |
organismQuantity |
Number of reads of this sequence variant in the sample | Number of positive droplets/chambers in the sample | Highly recommended |
organismQuantityType |
Should always be DNA sequence reads | The partition type | Highly recommended |
sampleSizeValue |
Total number of reads in the sample. This important since it allows calculating the relative abundance of the sequence variant within the sample | The number of accepted partitions (n), e.g. meaning accepted droplets in ddPCR or chambers in dPCR. | Highly recommended |
sampleSizeUnit |
Should always be DNA sequence reads | The partition type, should be equal to the value in organismQuantityType | Highly recommended |
materialSampleID |
An identifier for the MaterialSample (as opposed to a particular digital record of the material sample). Use the biosample ID if one was obtained from a nucleotide archive. In the absence of a persistent global unique identifier, construct one from a combination of identifiers in the record that will most closely make the materialSampleID globally unique. | An identifier for the MaterialSample (as opposed to a particular digital record of the material sample). Use the biosample ID if one was obtained from a nucleotide archive. In the absence of a persistent global unique identifier, construct one from a combination of identifiers in the record that will most closely make the materialSampleID globally unique. | Highly recommended |
samplingProtocol |
The name of, or reference to, or description of the method or protocol used during the sampling event. | The name of, or reference to, or description of the method or protocol used during the sampling event. | Recommended |
taxonID |
For eDNA data, it is recommended to use an MD5 hash of the sequence and prepend it with ASV. | metabarcoding: Highly recommended if DNA_sequence is not provided. for qPCR data, this field is not required. | |
scientificName |
Latin name of the closest known taxon (species or higher) or an OTU identifier from BOLD or UNITE | metabarcoding: not required. qPCR: Required |
After populating the Occurrence Core or extension with the (additional) required or highly recommended fields, additional information related to either your metabarcoding or qPCR data is to be included in the DNA-derived data extension. The (highly) recommended fields to include in this extension depend on whether your output is the result of metabarcode or qPCR analysis. The tables below only indicate the highly recommended or required fields - additional field should be included if applicable (see GBIF DNA-derived Data Extension Manual).
DNA-derived Data Extension Terms for Metabarcoding Data
Darwin Core Term Description Required DNA_sequenceThe DNA sequence (ASV). Highly recommended sopStandard operating procedures used in assembly and/or annotation of genomes, metagenomes or environmental sequences (e.g. through protocols.io) Recommended target_geneTargeted gene or marker name for marker-based studies Highly recommended target_subfragmentName of subfragment of a gene or markerImportant to e.g. identify special regions on marker genes like the hypervariable V6 region of the 16S rRNA gene Highly recommended pcr_primer_forwardForward PCR primer that was used to amplify the sequence of the targeted gene, locus or subfragment. Highly recommended pcr_primer_reverseReverse PCR primer that was used to amplify the sequence of the targeted gene, locus or subfragment. Highly recommended pcr_primer_name_forwardName of the forward PCR primer Highly recommended pcr_primer_name_reverseName of the reverse PCR primer Highly recommended pcr_primer_referenceReference for the primers Highly recommended
DNA-derived Data Extension Terms for qPCR Data
Darwin Core Term Description Required sopStandard operating procedures used in assembly and/or annotation of genomes, metagenomes or environmental sequences. A reference to a well documented protocol, e.g. using protocols.io Highly recommended annealingTempThe reaction temperature during the annealing phase of PCR. Required if annealingTemp was supplied annealinTempUnitHighly recommended pcr_condDescription of reaction conditions and components of PCR in the form of “initial denaturation:94degC_1.5min; annealing=…” Highly recommended probeReporterType of fluorophore (reporter) used. Probe anneals within amplified target DNA. Polymerase activity degrades the probe that has annealed to the template, and the probe releases the fluorophore from it and breaks the proximity to the quencher, thus allowing fluorescence of the fluorophore. Highly recommended probeQuencherType of quencher used. The quencher molecule quenches the fluorescence emitted by the fluorophore when excited by the cycler’s light source as long as fluorophore and the quencher are in proximity, quenching inhibits any fluorescence signals. Highly recommended ampliconSizeThe length of the amplicon in basepairs Highly recommended thresholdQuantificationCycleThreshold for change in fluorescence signal between cycles qPCR: Highly recommended baselineValueThe number of cycles when fluorescence signal from the target amplification is below background fluorescence not originated from the real target amplification. qPCR: Highly recommended
It is recommended that if ASVs are provided, MD5s should be generated by the biodiversity discovery platforms. If ASVs are not provided, the MD5s need to be mandatory. This will help data platform index actual sequences, or at the very minimum a MD5 checksum of these to facilitate searches for ASVs across datasets.
References
- Andersson AF, Bissett A, Finstad AG, Fossøy F, Grosjean M, Hope M, Jeppesen TS, Kõljalg U, Lundin D, Nilsson RN, Prager M, Svenningsen C & Schigel D (2021) Publishing DNA-derived data through biodiversity data platforms. v1.0 Copenhagen: GBIF Secretariat. https://doi.org/10.35035/doc-vf1a-nr22.
- The OBIS Manual: Examples
- Schema for DNA-Derived Data Extension
- OBIS Webinar on Genetic Data 28 Oct 2021
Example Datasets
- 18S Monterey Bay Time Series: an eDNA data set from Monterey Bay, California, including years 2006, 2013 - 2016
- Passive eDNA collection enhances aquatic biodiversity analysis, Australia (2019)
Relevant R/Python packages
R:
- DADA2: processing sequences to ASVs and more
- Phyloseq: tool to import, store, analyze, and graphically display complex phylogenetic sequencing data after clustering
Python:
- https://github.com/biocore/biom-format
- https://entrezpy.readthedocs.io/en/master/